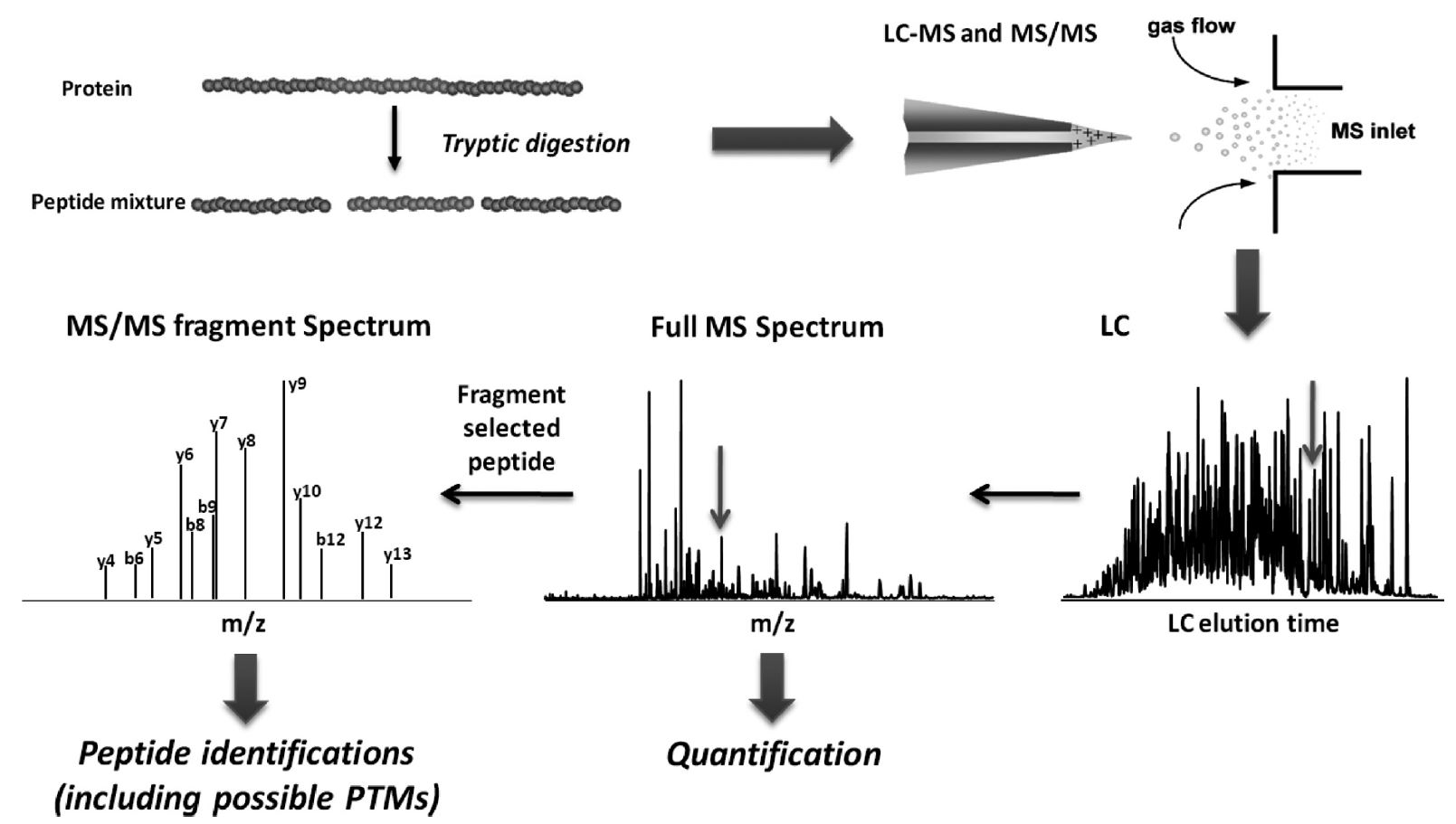
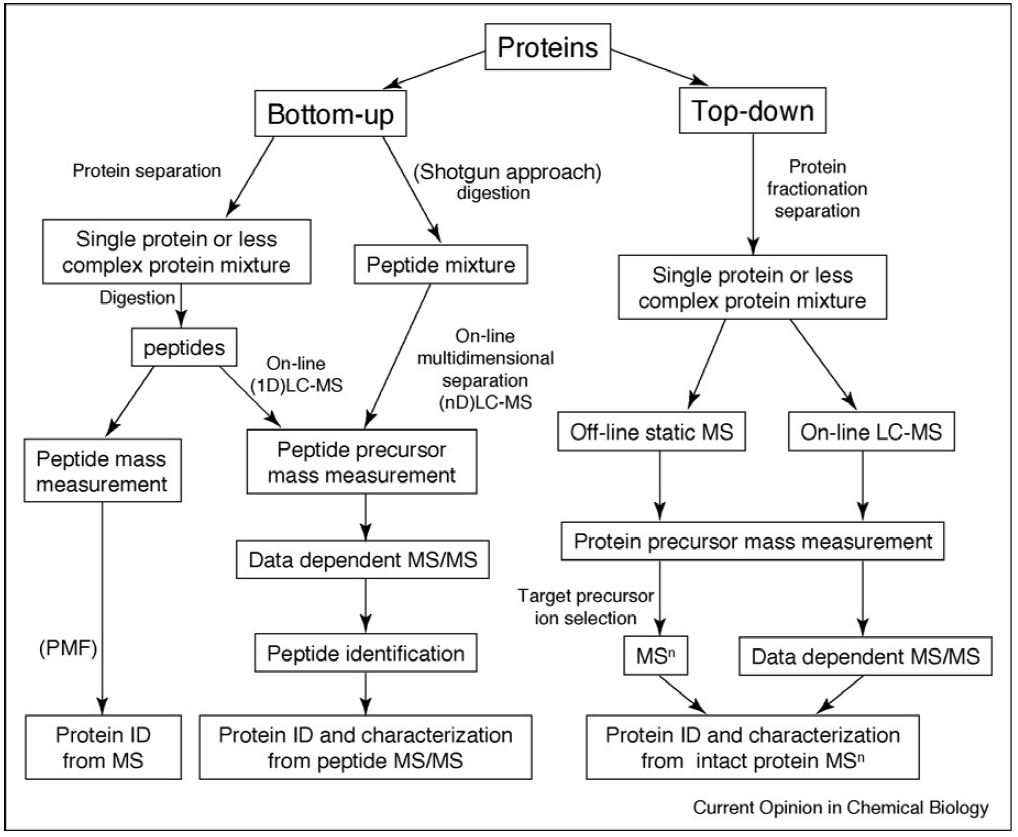
Strategies for MS-based protein identification and characterization. Proteins extracted from biological samples can be analyzed by bottom-up or top-down methods. In the bottom-up approach, proteins in complex mixtures can be separated before enzymatic (or chemical) digestion followed by direct peptide mass fingerprinting-based acquisition or further peptide separation on-line coupled to tandem mass spectrometry. Alternatively, the protein mixture can be directly digested into a collection of peptides (‘shotgun’ approach), which are then separated by multidimensional chromatography on-line coupled to tandem mass spectrometric analysis. In the top-down approach, proteins in complex mixtures are fractionated and separated into pure single proteins or less complex protein mixtures, followed by off-line static infusion of sample into the mass spectrometer for intact protein mass measurement and intact protein fragmentation. An on-line LC–MS strategy can also be used for large-scale protein interrogation.
Several
prominent mass spectrometry(MS)–based strategies are available:
• The most frequently used strategy
is referred to as shotgun or discovery proteomics. This strategy results in
datasets that can identify vast numbers of proteins contained in biological
samples but is more likely to reproducibly detect the most abundant proteins.
• In directed proteomics, specific,
predetermined segments of a proteome are identified and quantified at a higher
level of reproducibility, compared to discovery proteomics.
• Targeted proteomics generates
highly reproducible and accurate datasets of a small, preselected fraction of a
proteome (typically one to a few hundred peptides) with high detection
sensitivity and dynamic range.
• With recent advances in instrumentation, a fourth strategy referred to as data-independent analysis (DIA) is emerging in which the identification of all proteins is attempted for each sample.